- ホーム
- 研究紹介








【お問い合わせ】
〒860-0811
熊本市中央区本荘2-2-1
熊本大学
生命資源研究・支援センター
分子血管制御分野
TEL / FAX : 096-373-6500

研究紹介
血管動態の表現型解析研究から、がん・動脈硬化・血栓症などの血管の病態を理解し、治療法を考える
現在の超高齢化社会を迎え、脳卒中・心筋梗塞の素因となり血栓症や動脈硬化症及び病的な血管新生に起因するがん増殖・転移での死亡率は年々上昇しています。これらの病態にはいずれも血管が深く関与しており、血管の生理・病理変化に焦点をおき、凝固・炎症・透過性・血管新生の基本原理を分子レベルで解明していくことがその第一ステップです。特に血管系の基礎を構築する内皮細胞での遺伝子発現変化・エピゲノム変化を包括的に追跡し、かつその制御システムを理解していくことに挑戦していきます。
アプローチ1:血管内皮細胞の動態変化を包括的に調べる
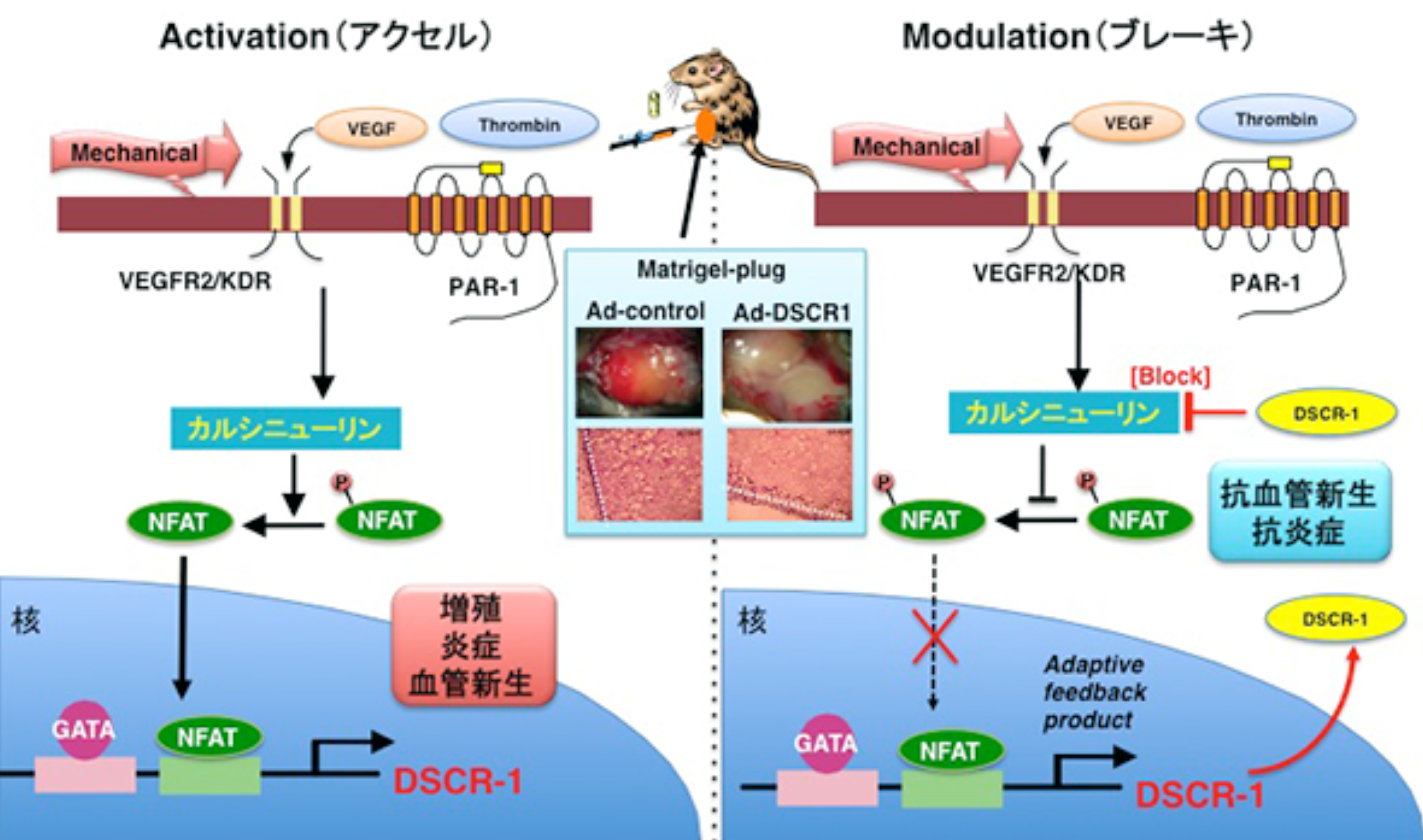
~アクセルとブレーキを介した内皮活性化システム~
内皮恒常性や血管構築に強く寄与する VEGF や血栓に関与するトロンビンは転写因子 NFAT の核内移行や EGR3 誘導を行う代表的な内皮アクセルであり、血管新生に必須な各因子の発現誘導を行うが、恒常的に強く誘導し続けるとアポトーシス関連因子の誘導を介して内皮自体が不安定化する。常に適切なフィードバック系路がないと恒常性の維持が出来ない仕組みとなっている。EGR3 はそのフィードバック系として、NAB2 タンパクを誘導し、一方 NFAT は上流のカルシニューリンを特異的にフィードバック調節する因子として私たちはダウン症因子 (DSCR)-1 を見出している (Minami, et.al. 2004, 2006 JBC)。これらブレーキシステムは活性化シグナルを適切に伝達し、多くの因子の相互作用によって成り立つ血管新生を正常に進めるのに必須である。DSCR-1 のノックアウトマウスは NFAT 活性化が過剰で、透過性亢進並びに胎生期における部分的な脳血管の出血を呈する (Ryeom et.al. Cancer cell 2007)。さらに炎症度が構成的に高く、敗血症などの急性期ストレスに脆弱となる (Minami et.al. J.Clin.Invest. 2009)。また血管密度に比して VEGF 濃度が高い腫瘍原発巣においては逆に血管新生が抑制される結果となる (Minami et.al. Cell Rep.2013)。その一方で DSCR-1 の構成的発現トランスジェニックマウスもアクセル/ブレーキでの閉じた系(恒常性)を破綻させる。DSCR-1 遺伝子座 Bac トランスジェニック(Tg)マウスは胎生致死であることは知られており、筆者らの内皮特異的 DSCR-1 コンディショナル Tg マウスにおいても DSCR-1 ブレーキの発現量が多いと、血管総数減少による発育不全や血管分岐異常を呈する。しかしながらブレーキを効率良く、かつタイミング良くかけることによって病的な内皮活性化を抑制することも可能である。固形がん、メラノーマの癌種を移植した xenograft マウスでのがん増殖は、血管新生を強く抑制することに基づいて大きく遅延し、その炎症度や最終的な生存度も改善する。ダウン症患者が固形がんにかかりにくい疫学の論理もDSCR-1の発現度に大きく依存していることも私たちの国際共同研究から明らかとなっている (Baek et.al. Nature 2009)。このようにフィードバック因子の発現量のバランスによって大きく表現型が異なるが、シグナル制御の中心を担うこのようなブレーキ因子は今後の新たな抗血管新生創薬としての価値が見出される可能性がある。
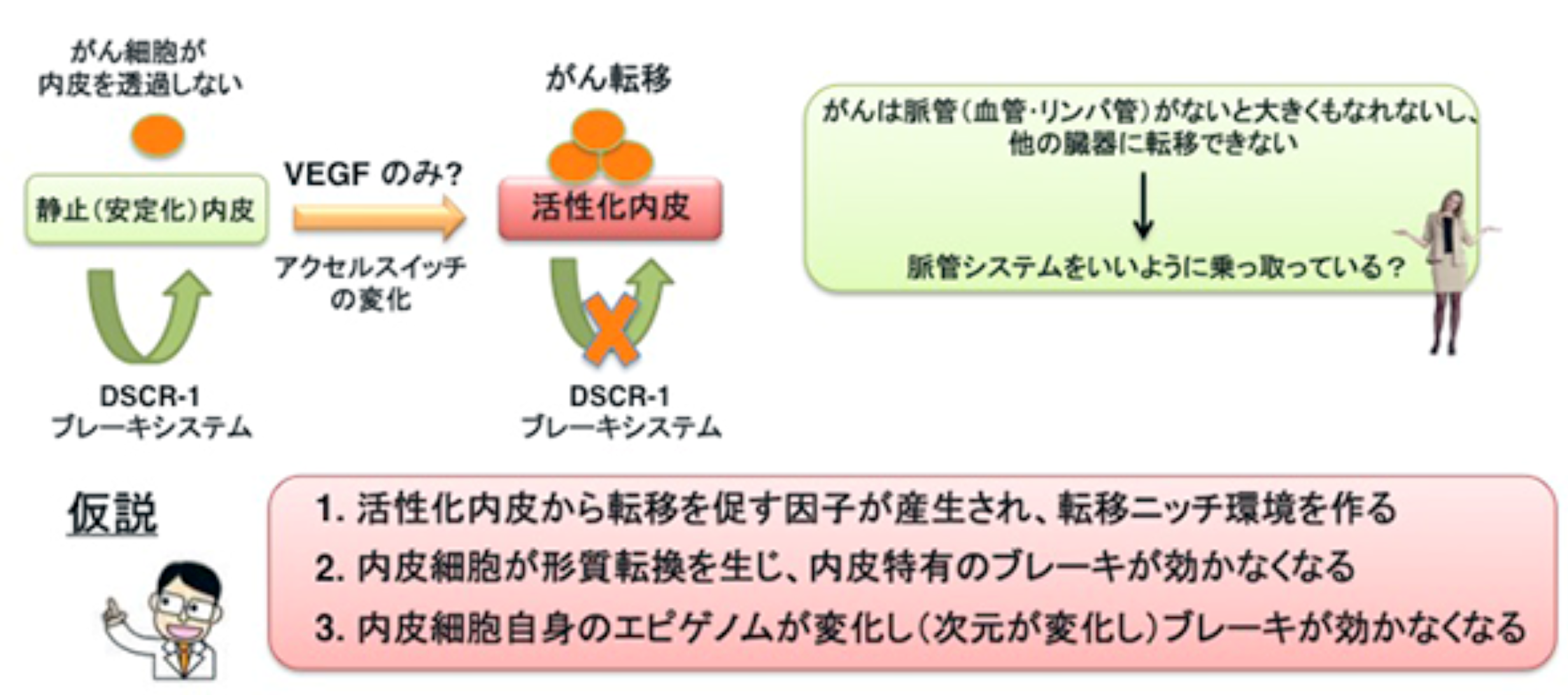
アプローチ2:恒常性システムの破綻による血管病の分子機構を解明する
生体は優れた恒常性維持システムを保有しており、血管内皮細胞も様々な刺激やストレスをアクセル/ブレーキシステムを介して下流に適切に伝え、血流・血圧・自然免疫・炎症や凝固・血管新生反応を担っている (Minami J. Biochem. review 2014)。そのシステムの破綻が病的な活性化に繋がると想定されるが、DSCR-1 ブレーキシステムを考慮した場合、通常 2 コピー存在しているのに、一旦がんが形成されると、無処置の場合大きく増殖し、また他臓器へ血管やリンパ管を通して転移する。あらかじめブレーキの量を増やしておくとがん転移は遅延するが、完全に防護はできない。即ち、ブレーキシステムが効かない微小環境に陥っていることが想定される。転移は血管・リンパ管などの管を通して必ず引き起こされるので、内皮活性化に変化が生じた可能性が示唆される。私たちは、その可能性として、① 1つの活性化シグナルによって 新たな因子が内皮から分泌され、転移が進む。②病態微小環境下、内皮細胞の形態や内皮特異性が変化し、内皮特有のブレーキが効かなくなる。③ 内皮細胞においてエピゲノム変化が生じ、自己終息しなくなる。これら3つの仮説を考えている。
まず、1番目の新しい環境要因が加わる可能性であるが、例えば VEGF シグナルに2型ヘルパー T 細胞が主に分泌する慢性的な IL-4/13シグナルが加わると、持続的な炎症反応となり、VEGF によって引き起こされる内皮への単球接着反応(炎症初期反応)も DSCR-1 安定発現のみでは終息しなくなる (Tozawa. et.al. Mol.Cell.Biol. 2011)。がん細胞においても同じように血管内に侵入する過程にこのような慢性シグナルが関与している可能性が示唆される。また私たちは近年、NFAT の下流で血管新生性マクロファージの動員や内皮不安定化に寄与する Angiopoietin (ANG)-2 を見出している (Minami et.al. Cell Rep. 2013)。次に2番目の可能性であるが、心弁形成時や病的な梗塞時・がん微小環境下において、血管内皮細胞が間葉系細胞様の性質に変化する Endothelial cell-mesenchymal transition (EndMT) という事象が起きる可能性について近年考えられている。
さらに内皮分化を運命付ける転写因子カスケードについても明解になってきている。このカスケードの末端に位置する2つの ETS 因子が GATA2 の影響を受けて安定発現し、内皮を規定しているが (Kanki et.al. EMBO J.2011)、がん微小環境下においてこれらの転写因子の発現が下がり、EndMT を引き起こすきっかけとなっていることを免疫染色やゲノムワイド ChIP-seq から解明途中である。最後に3番目の可能性であるが、VEGF 刺激における網羅的エピゲノムマッピングから、血管新生に必須な転写因子群の発現制御には必ず H3K4me3 ヒストン修飾の増大が生じており、そのアクセルマークを入れるトリソラックス複合体をクロマチンに動員するアクセサリータンパクが NFAT と相互作用して核内移行し、標的配列のクロマチン修飾に関与している可能性を現在調査している。このアクセサリータンパクを発現阻害すると、内皮恒常性は維持したまま VEGF 刺激におけるアクセルスイッチを切ってしまうので、病的な血管新生やがん増殖は大きく抑制される結果となる。これら3つの可能性は VEGF 阻害剤における抗腫瘍血管阻害法単独では成功しなかった VEGF 非依存性獲得やがん悪性化、転移能亢進を抑制しうる新たな方法論であり、タンパク相互作用阻害剤などの開発を通じた創薬への発展も期待したい。